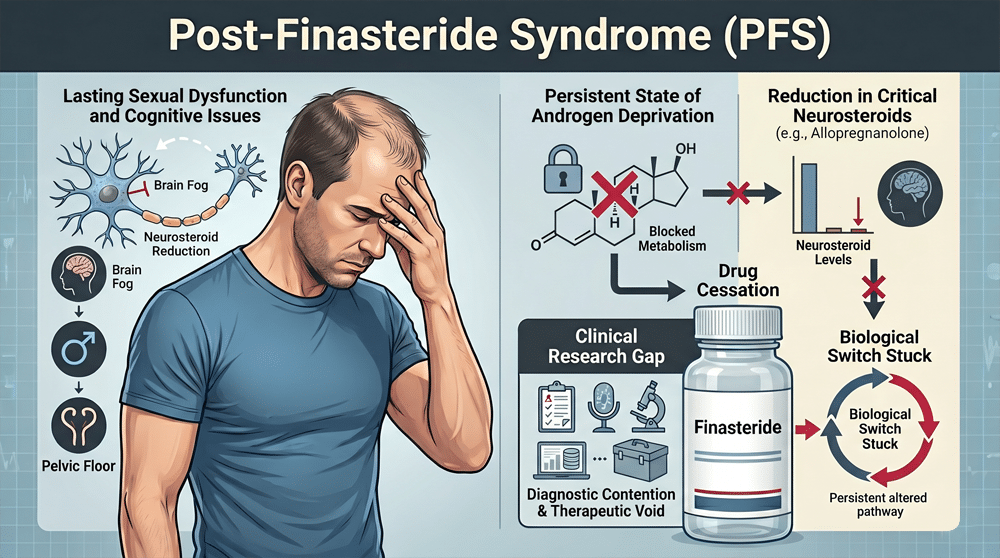

The 5-Alpha Reductase Inhibition Cascade: Post-Drug Physiological Failure
Finasteride and dutasteride function as competitive inhibitors of 5-alpha reductase enzymes. These medications block the conversion of testosterone to dihydrotestosterone; this mechanism treats benign prostatic hyperplasia and androgenetic alopecia. The clinical fallout occurs when the medication is stopped but the physiological system fails to recalibrate to its baseline state.
The pharmacological intervention alters neurosteroid synthesis pathways; it’s this specific enzymatic disruption that’s linked to the persistent clinical phenotype known as post-finasteride syndrome or PFS. This syndrome isn’t merely a lack of androgenic drive; it’s a structural and epigenetic shift in neural signaling. Patients often report symptoms that are completely refractory to standard hormone replacement therapy.
5-alpha reductase exists as three distinct isoenzymes with tissue-specific distributions. Type 1 predominates in skin and liver; type 2 localizes to prostate and genital skin. Type 3 appears in brain tissue and contributes significantly to the production of critical neurosteroids.
The inhibition extends well beyond peripheral androgen metabolism.
Neurosteroids including allopregnanolone and tetrahydrodeoxycorticosterone require 5-alpha reductase for synthesis. These compounds modulate GABA-A receptor function and influence mood regulation through established neuroendocrine pathways.
Neurosteroid Depletion and GABA-A Receptor Dysfunction
Allopregnanolone acts as a potent positive allosteric modulator of GABA-A receptors. This neurosteroid enhances chloride ion conductance; the resulting hyperpolarization produces anxiolytic and sedative effects. Depletion creates a functional GABA deficit.
Chronic 5-alpha reductase inhibition reduces allopregnanolone synthesis in the brain. Cerebrospinal fluid measurements confirm decreased concentrations; the magnitude correlates with symptom severity. Clinical studies demonstrate higher rates of neuropsychiatric symptoms in former users.
GABA-A receptor subunit composition changes in response to chronic neurosteroid deprivation. Alpha and gamma subunit expression shifts; this alters receptor pharmacology and benzodiazepine sensitivity. The neuroadaptation explains persistent symptoms despite medication cessation.
The Post-Finasteride Syndrome Clinical Phenotype
Post-Finasteride Syndrome encompasses persistent side effects following drug discontinuation. Sexual dysfunction manifests as libido loss and erectile impairment; these symptoms often prove refractory to standard treatments. The androgen-independent nature distinguishes PFS from hypogonadism.
Cognitive symptoms include memory impairment and executive dysfunction. Patients report mental fog and reduced processing speed; formal neuropsychological testing confirms deficits in attention and working memory. Neuroprotection strategies address these changes.
Mood disturbances range from anxiety to major depression. The severity exceeds expected rates for the underlying conditions treated; this suggests a pharmacological etiology. GABA modulation may provide symptomatic relief.
Dopaminergic and Serotonergic System Disruption
Neurosteroids influence monoamine neurotransmitter systems beyond GABA modulation. Allopregnanolone affects dopamine release in mesolimbic pathways; depletion may contribute to anhedonia and motivational deficits. Dopaminergic support warrants investigation.
Serotonergic signaling also responds to neurosteroid modulation. 5-HT1A receptor function changes with allopregnanolone levels; this mechanism links to mood and sexual function. The interaction explains overlapping symptoms with SSRI discontinuation syndromes.
Chronic inhibition may alter gene expression in neural tissue. Epigenetic modifications affecting steroidogenic enzymes persist; this molecular memory explains symptom persistence. Sleep architecture disruption compounds these effects.
Neuroendocrine Dysregulation Beyond Androgens
The hypothalamic-pituitary-gonadal axis shows complex alterations in PFS. Testosterone levels often remain within normal ranges; the androgen receptor sensitivity appears compromised. This androgen resistance pattern differs from primary hypogonadism.
Corticotropin-releasing hormone dynamics change with chronic neurosteroid depletion. The stress response system demonstrates dysregulation; HPA axis function requires careful assessment. Cortisol patterns may show flattening or inappropriate elevation.
Thyroid function tests warrant evaluation in PFS patients. Clinical overlap with hypothyroidism exists; metabolic symptoms compound the primary condition. Thorough endocrine profiling guides individualized treatment.
Androgen Receptor Dynamics and Tissue Resistance
Androgen receptor expression changes in response to chronic 5-alpha reductase inhibition. Receptor density may increase as a compensatory mechanism; this upregulation persists after drug cessation. The tissue-level androgen resistance complicates hormone replacement approaches.
Peripheral tissues demonstrate variable sensitivity to circulating testosterone. Despite normal serum levels, target organs experience functional hypogonadism. This discordance between laboratory values and clinical symptoms frustrates conventional diagnostic approaches.
Genetic polymorphisms in androgen receptor genes may influence susceptibility. CAG repeat length variations affect receptor sensitivity; longer repeats correlate with reduced transcriptional activity. Pharmacogenetic factors explain individual differences in PFS development.
Peripheral and Central Nervous System Manifestations
Penile tissue changes include altered vascular responsiveness and fibrosis. Histological studies demonstrate collagen deposition; endothelial function appears compromised. These structural changes contribute to persistent erectile dysfunction.
Prostatic tissue undergoes atrophy with chronic inhibition. The intended therapeutic effect becomes pathological when persistent; glandular function remains suppressed. Urinary symptoms may paradoxically worsen or persist despite treatment indication.
Skin and hair follicles show altered physiology. Sebaceous gland activity decreases; wound healing may slow. The cosmetic benefits of treatment reverse partially but incompletely following discontinuation.
Neuroinflammation and Oxidative Stress Mechanisms
Neurosteroid depletion may trigger inflammatory cascades in neural tissue. Microglial activation increases with chronic allopregnanolone deprivation; proinflammatory cytokine levels rise. The neuroinflammatory state contributes to cognitive and mood symptoms.
Oxidative stress markers elevate in PFS patients. Lipid peroxidation and protein oxidation indicate cellular damage; antioxidant defenses appear compromised. Mitochondrial dysfunction may drive this oxidative burden.
The blood-brain barrier integrity warrants investigation in Post Finasteride Syndrome. Neuroinflammation may compromise endothelial tight junctions; increased permeability could amplify CNS exposure to systemic factors. Neuroprotective strategies should address barrier function.
Treatment Strategies and Recovery Protocols
Neurosteroid restoration represents a primary therapeutic target. Allopregnanolone analogs and precursors show preliminary efficacy; synthetic neurosteroids bypass the blocked synthesis pathway. Clinical trials remain limited but promising.
Hormonal interventions require careful titration and monitoring. Testosterone replacement alone often proves insufficient; combination approaches addressing neurosteroid and androgen pathways may prove superior. Individual response varies widely.
Psychiatric symptom management addresses the secondary mood and cognitive effects. Selective serotonin reuptake inhibitors demonstrate partial efficacy; novel agents targeting neurosteroid-sensitive receptors warrant investigation. Supportive care remains critical.
Prognosis and Long-Term Outcomes
The natural history of Post-Finasteride Syndrome remains poorly characterized. Some patients report gradual improvement over months to years; others experience persistent symptoms for decades. The variable course complicates treatment efficacy assessment.
Recovery appears more likely in cases with shorter drug exposure duration. Younger patients may demonstrate greater neural plasticity; this resilience supports functional recovery. Early intervention likely improves outcomes.
Research priorities include biomarker identification and mechanistic studies. Validated outcome measures would enable clinical trials; pathophysiological understanding would guide targeted therapies. Patient registries contribute to natural history data collection.
SRD5A2 Epigenetic Silencing and Persistent Gene Dysregulation
The SRD5A2 gene encoding 5-alpha reductase type II demonstrates altered methylation patterns in PFS patients. CpG island hypermethylation at promoter regions suppresses transcriptional activity; this epigenetic modification persists after drug discontinuation. The molecular memory explains the syndrome’s refractory nature.
Histone modifications accompany DNA methylation changes in affected tissues. Histone deacetylation and methylation create a repressive chromatin environment; these marks alter gene expression profiles. The epigenetic landscape shifts toward reduced steroidogenic capacity.
Transcription factor binding at SRD5A2 promoters changes with chronic inhibition. Androgen receptor and other nuclear factors demonstrate altered recruitment; the regulatory network adapts to persistent drug exposure. These changes may not reverse spontaneously.
Neuroactive Steroid Signaling and GABA-A Receptor Reorganization
The progesterone-to-allopregnanolone pathway represents a critical neurosteroid synthetic route. Pregnenolone converts to progesterone; subsequent 5-alpha reduction produces allopregnanolone. 5-alpha reductase inhibition blocks this terminal step.
GABA-A receptor subunit composition reorganizes in response to chronic allopregnanolone depletion. Delta and gamma subunit expression changes alter receptor pharmacology; the extrasynaptic receptor population particularly affected. Neurosteroid-sensitive receptors decrease in density.
Alpha-4 and beta-2 subunit upregulation occurs as a compensatory mechanism. These subunits confer different kinetic properties; the receptors demonstrate reduced sensitivity to GABA and neurosteroids. The molecular adaptation parallels changes seen in alcohol withdrawal.
Phasic and tonic inhibition patterns shift with receptor reorganization. Synaptic GABA transmission remains partially intact; extrasynaptic tonic inhibition suffers greater impairment. The imbalance affects network excitability and seizure threshold.
Mitochondrial Bioenergetics and Oxidative Metabolism
Neurosteroids influence mitochondrial membrane potential through direct and indirect mechanisms. Allopregnanolone stabilizes the inner mitochondrial membrane; depletion increases proton leak and reduces coupling efficiency. Dopaminergic support can’t fully compensate for this energetic deficit.
Oxidative phosphorylation capacity decreases in neural tissue with chronic neurosteroid deprivation. Complex I and Complex IV activities show modest reductions; ATP production fails to meet peak metabolic demands. Mitochondrial protection strategies address this vulnerability.
Reactive oxygen species generation increases relative to antioxidant defenses. Superoxide dismutase and catalase activities may not keep pace; oxidative damage accumulates in mitochondrial DNA and membranes. The vicious cycle of dysfunction perpetuates cellular injury.
Calcium handling by mitochondria changes with membrane potential alterations. The organelle’s ability to sequester calcium during excitatory events diminishes; cytosolic calcium rises and triggers degenerative cascades. Sleep quality affects these calcium dynamics.
HPA Axis Dysregulation and Stress System Pathology
The hypothalamic-pituitary-adrenal axis demonstrates complex dysregulation in PFS. Corticotropin-releasing hormone neurons show altered activity; the central stress response system becomes sensitized. Chronic allopregnanolone deprivation removes a key modulatory influence.
Glucocorticoid receptor sensitivity changes with chronic neurosteroid depletion. Receptor expression and function may downregulate; this alters negative feedback and prolongs stress responses. The neuroendocrine system loses flexibility.
Circadian cortisol patterns flatten or phase-shift in affected patients. Morning cortisol peaks may attenuate; evening nadir elevation impairs sleep onset. The dysrhythmia compounds mood and cognitive symptoms.
5-Alpha Reductase Inhibitors Technical Specifications
| Parameter | Finasteride | Dutasteride |
|---|---|---|
| Isoenzyme Selectivity | Type 2 selective | Type 1 and 2 dual |
| Half-life | 6-8 hours | 5 weeks (terminal) |
| Volume of Distribution | 76 liters | 300-500 liters |
| Brain Penetration | Limited (BBB restricts) | Limited (BBB restricts) |
| 5-ARI Dissociation | Reversible | Slow off-rate |
Comparative Analysis: Inhibition Strategies
| Compound | Tissue-Specific Inhibition | BBB Permeability |
|---|---|---|
| Finasteride | Prostate/Genital skin (Type 2) | Poor; peripheral action predominant |
| Dutasteride | Systemic (Types 1 and 2) | Poor; longer tissue retention |
| Saw Palmetto | Weak/Variable inhibition | Minimal; lipophilic extracts |
The 2026 Neurosteroid Support Stacking Protocol
| Target System | Primary Compound | Supportive Stack | Protocol Rationale |
|---|---|---|---|
| GABA-A Modulation | Allopregnanolone Analogs | Agmatine Sulfate | NMDA modulation complements GABA |
| Mitochondrial Support | PQQ | P-5-P (B6) | Neurotransmitter synthesis support |
| Dopaminergic Tone | Bromantane | L-Tyrosine | Tyrosine hydroxylase upregulation |
| Sleep Architecture | Glycine | Magnesium L-Threonate | NMDA modulation + GABA enhancement |
| HPA Axis Regulation | Phosphatidylserine | Ashwagandha | Cortisol response modulation |
Clinical Decision Framework for Recovery
The restoration of neurosteroid signaling represents the primary therapeutic target. Synthetic allopregnanolone analogs bypass the blocked synthesis pathway; these agents show promise in preliminary studies. Regulatory approval for PFS indications remains pending.
Hormonal interventions require careful monitoring and individualized dosing. Testosterone replacement alone rarely suffices; combination approaches addressing multiple pathways may prove superior. Allopregnanolone in mood disorders provides mechanistic guidance.
Neuroprotective strategies support cellular recovery during treatment. Mitochondrial support compounds, antioxidants, and anti-inflammatory agents address downstream consequences. The multimodal approach reflects the syndrome’s complexity.
Sexual Dysfunction Mechanisms Beyond Androgen Deprivation
Penile tissue undergoes structural changes in Post-Finasteride Syndrome that resist androgen replacement. Collagen deposition increases within the corpus cavernosum; this fibrosis reduces tissue compliance. The architectural changes impair hemodynamic responses.
Endothelial dysfunction in penile vasculature contributes to erectile impairment. Nitric oxide synthesis and signaling show reduced capacity; the relaxation response to neural stimulation attenuates. Vascular insufficiency compounds the neurological deficits.
Genital sensation changes reflect peripheral nerve alterations. Small fiber neuropathy may develop; reduced tactile and thermal sensitivity impairs sexual response. The sensory pathway disruption explains pleasure deficits independent of hormonal status.
Cognitive Domain Deficits and Neural Network Disruption
Working memory impairments in PFS correlate with hippocampal neurosteroid depletion. Allopregnanolone supports synaptic plasticity in CA1 and CA3 regions; chronic deprivation impairs long-term potentiation. Sleep disruption compounds these deficits.
Executive function tests demonstrate reduced cognitive flexibility and set-shifting. Prefrontal cortex-dependent processes show vulnerability; the neurosteroid modulation of GABAergic interneurons affects cortical microcircuit function. Attentional control suffers measurable impairment.
Processing speed reductions appear on standardized neuropsychological batteries. Simple and choice reaction times increase; the slowing affects multiple cognitive domains. Motor cortex excitability changes may contribute to these findings.
Mood and Anxiety Symptom Complex
Anhedonia represents a core affective symptom in Post Finasteride Syndrome patients. The loss of pleasure affects sexual, social, and aesthetic domains; this pervasive deficit suggests mesolimbic pathway involvement. Dopaminergic restoration attempts address this target.
Generalized anxiety manifests independently of situational triggers. The GABA-A receptor hypoactive state generates baseline arousal; patients describe persistent unease and vigilance. Benzodiazepine sensitivity changes complicate pharmacological management.
Depressive episodes may meet diagnostic criteria for major depressive disorder. The neurovegetative symptoms include sleep and appetite changes; cognitive distortions affect self-perception and hopelessness. Standard antidepressants demonstrate variable efficacy.
Peripheral Tissue Manifestations
Muscle tissue shows altered composition and function in PFS. Type II fiber atrophy may occur; strength and power generation decrease despite exercise. The myopathic changes resist androgen replacement alone.
Adipose tissue distribution shifts with chronic 5-alpha reductase inhibition. Subcutaneous fat may increase while visceral patterns change; metabolic effects include altered lipid profiles. Insulin sensitivity warrants monitoring.
Bone mineral density requires assessment in long-term PFS. The role of dihydrotestosterone in skeletal maintenance remains underappreciated; osteopenia may develop insidiously. Dual-energy x-ray absorptiometry provides baseline data.
Biomarker Development and Diagnostic Challenges
Objective biomarkers for PFS remain under development. Neurosteroid measurements in cerebrospinal fluid show promise; peripheral blood levels correlate poorly with CNS status. The invasive sampling limits routine clinical application.
Genetic screening for susceptibility variants may guide prevention strategies. Polymorphisms in SRD5A2, AR, and neurosteroid synthetic enzymes affect risk; pharmacogenetic testing could identify vulnerable individuals. Epigenetic assays remain research tools.
Functional neuroimaging reveals patterns distinct from primary psychiatric disorders. Hippocampal and prefrontal metabolism shows characteristic changes; these findings support the organic nature of cognitive symptoms. Longitudinal studies track recovery trajectories.
Emerging Therapeutic Approaches
Neurosteroid replacement therapy represents the most direct mechanistic approach. Synthetic allopregnanolone analogs bypass the blocked synthetic pathway; clinical trials assess safety and efficacy. Regulatory pathways for PFS indications require development.
Epigenetic modification strategies target the persistent gene dysregulation. Histone deacetylase inhibitors and DNA demethylating agents show preclinical promise; translation to human therapy requires careful safety evaluation. The reversibility of epigenetic marks offers hope.
Regenerative medicine approaches explore stem cell and exosome therapies. Neural progenitor cell transplantation aims to restore neurosteroid synthetic capacity; early-phase trials assess feasibility. The invasive nature limits initial application to severe cases.
Patient Advocacy and Research Priorities
The PFS patient community drives research awareness and funding. Online forums document the syndrome’s scope; advocacy organizations engage with regulatory agencies. The grassroots movement accelerates scientific recognition.
Clinician education remains a critical need. Medical curricula rarely address PFS; continuing education programs fill gaps in knowledge. Specialist referral networks develop expertise in management.
Research priorities include mechanistic studies and clinical trials. Natural history data inform prognosis; treatment trials establish evidence-based practice. International collaboration advances understanding of this global condition.
Regulatory Considerations and Risk Communication
Drug regulatory agencies have updated product labeling for 5-alpha reductase inhibitors. The warnings now include persistent sexual dysfunction and mood changes; post-marketing surveillance continues to identify adverse events. Informed consent requires discussion of these risks.
Prescribing practices evolve with emerging safety data. Dermatologists and urologists reconsider risk-benefit ratios for cosmetic indications; alternative treatments gain consideration. Patient autonomy demands full disclosure of uncertainty.
Legal frameworks address liability for drug-induced harm. Litigation establishes precedent for informed consent standards; settlements fund research and patient support. The evolving legal landscape shapes clinical practice.
Future Directions and Clinical Translation
Pathophysiology of Post-Finasteride Syndrome continues to clarify with ongoing research. Mechanistic understanding advances therapeutic development; targeted interventions emerge from molecular insights. The clinical community increasingly recognizes this condition.
Multidisciplinary care models optimize patient outcomes. Endocrinology, neurology, psychiatry, and sexual medicine collaboration addresses the syndrome’s complexity. Coordinated care improves quality of life.
Prevention strategies may reduce future case incidence. Informed consent practices improve; pharmacogenetic screening identifies susceptible individuals. The tragedy of PFS drives safer therapeutic development.
Evidence guides clinical progress.
Clinical precision remains paramount.
Clinical Anecdotes & Human Biohacking Experience
The data tells one story. Real people tell another. These aren’t clinical trial participants; they’re biohackers, patients, Reddit users, and self-experimenters who’ve documented their PFS journeys.
“I took finasteride for minor hair loss and started having panic attacks. I stopped and they went away; however, this drug is often associated with symptom clusters of this nature and the drug itself has warning labels of such.” — u/chmpgne
“In spite of a modest increase in testosterone, patients being treated with Finasteride will often experience symptoms of androgen deprivation such as sexual dysfunction, depression, and cognitive issues.” — u/AccutaneEffectsInfo
“The cause of this condition, referred to as Post Finasteride Syndrome (PFS), remains elusive to the medical community.” — u/AccutaneEffectsInfo
“A study of rats found that one month following treatment with Finasteride there was a significant change to the composition of the gut microbiome.” — u/AccutaneEffectsInfo
“Another study on rats found that sub chronic treatment with finasteride reduced the gut concentrations of a variety of steroids including DHT and Allopregnanolone.” — u/AccutaneEffectsInfo
Raw experience matters. It isn’t controlled or pretty. But it’s real.
The SuperMindHacker Clinical Assessment
Post-Finasteride Syndrome represents a complex neuroendocrine disorder with persistent symptomatology. The 5-alpha reductase inhibition mechanism disrupts multiple pathways beyond peripheral androgen metabolism. Neurosteroid depletion emerges as a central pathophysiological factor.
Clinical management requires multidisciplinary approaches addressing hormonal, neurological, and psychiatric dimensions. Evidence-based interventions remain limited; case reports and mechanistic reasoning guide current practice. Research into neurosteroid restoration shows preliminary promise.
The recognition of PFS as a legitimate clinical entity continues evolving. Regulatory agencies have updated product labeling; medical education increasingly addresses this condition. Patient advocacy drives research funding and clinical attention to this underrecognized syndrome.

0 Comments